Théorie des acides et des bases. Théorie de Bronsted-Lowry. Acides et bases conjugués Théorie de Bronsted-Lowry. Acides et bases conjugués
Selon Lewis, les propriétés acides et basiques des composés organiques sont mesurées par la capacité d'accepter ou de donner une paire d'électrons, suivie de la formation d'une liaison. Un atome qui accepte une paire d'électrons est un accepteur d'électrons, et un composé contenant un tel atome doit être classé comme un acide. Un atome qui fournit une paire d'électrons est un donneur d'électrons, et un composé contenant un tel atome est une base.
Plus précisément, les acides de Lewis peuvent être un atome, une molécule ou un cation : proton, halogénures d'éléments des deuxième et troisième groupes du système périodique, halogénures de métaux de transition - BF3, ZnCl2, AlCl3, FeCl3, FeBr3, TiCl4, SnCl4, SbCl5, métal cations, anhydride sulfurique - SO3, carbocation. Les bases de Lewis comprennent les amines (RNH2, R2NH, R3N), les alcools ROH, les éthers ROR
Selon Bronsted-Lowry, les acides sont des substances capables de donner un proton et les bases sont des substances qui acceptent un proton.
Acide et base conjugués :
HCN (acide) et CN- (base)
NH3 (base) et NH4+ (acide)
L'équilibre acido-basique (ou protolytique) est un équilibre auquel participe un proton (H+).
HCOOH + H 2 O D H 3 O + + HCOO -
acide 2 base 1
H 2 O + NH 3 D NH 4 + + OH -.
acide 1 base 2 conjugué conjugué
acide 2 base 1
7. Types d'isomérie en chimie organique. Isomérie structurale, spatiale et optique. Chiralité. Compatibilité et configuration. R, S, Z, E - nomenclature.
Il existe deux types d'isomérie : structurelle et spatiale (stéréoisomérie). Les isomères structuraux diffèrent les uns des autres dans l'ordre des liaisons des atomes dans une molécule, les stéréo-isomères - dans la disposition des atomes dans l'espace avec le même ordre de liaisons entre eux.
Isomérie structurale : isomérie du squelette carboné, isomérie de position, isomérie de diverses classes de composés organiques (isomérie interclasse).
Isomérie structurale
Isomérie du squelette carboné
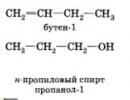
 L'isomérie de position est due à la position différente de la liaison multiple, du substituant, du groupe fonctionnel avec le même squelette carboné de la molécule :
L'isomérie de position est due à la position différente de la liaison multiple, du substituant, du groupe fonctionnel avec le même squelette carboné de la molécule :
Isomérie spatiale
L'isomérie spatiale est divisée en deux types : géométrique et optique.
L'isomérie géométrique est caractéristique des composés contenant des doubles liaisons et des composés cycliques. La libre rotation des atomes autour d'une double liaison ou dans un cycle étant impossible, les substituants peuvent être situés soit d'un côté du plan de la double liaison ou du cycle (position cis), soit sur des côtés opposés (position trans).
 L'isomérie optique se produit lorsqu'une molécule est incompatible avec son image dans un miroir. Ceci est possible lorsque l'atome de carbone de la molécule a quatre substituants différents. Cet atome est dit asymétrique.
L'isomérie optique se produit lorsqu'une molécule est incompatible avec son image dans un miroir. Ceci est possible lorsque l'atome de carbone de la molécule a quatre substituants différents. Cet atome est dit asymétrique.
CHIRALITÉ, propriété d'un objet d'être incompatible avec sa réflexion dans un miroir plan idéal.
Diverses structures spatiales qui surviennent en raison de la rotation autour de liaisons simples sans violer l'intégrité de la molécule (sans rompre les liaisons chimiques) sont appelées CONFORMATIONS.
La structure des alcanes. Sp3 est l'état du carbone. Caractérisation des liaisons C-C et C-H. Le principe de rotation libre. conformation. Modes de représentation et nomenclature. Propriétés physiques des alcanes.
 Tous les atomes de carbone dans les molécules d'alcane sont dans l'état hybridation sp 3, l'angle entre les liaisons C-C est de 109 ° 28 ", par conséquent, les molécules d'alcanes normaux avec un grand nombre d'atomes de carbone ont une structure en zigzag (zigzag). La longueur de la liaison C-C dans les hydrocarbures saturés est de 0,154 nm
Tous les atomes de carbone dans les molécules d'alcane sont dans l'état hybridation sp 3, l'angle entre les liaisons C-C est de 109 ° 28 ", par conséquent, les molécules d'alcanes normaux avec un grand nombre d'atomes de carbone ont une structure en zigzag (zigzag). La longueur de la liaison C-C dans les hydrocarbures saturés est de 0,154 nm
La liaison C-C est covalente non polaire. La liaison C-H est covalente et faiblement polaire, car C et H sont proches en électronégativité.
Propriétés physiques
Dans des conditions normales, les quatre premiers membres de la série homologue des alcanes sont des gaz, C 5 -C 17 sont des liquides et à partir de C 18 sont des solides. Les points de fusion et d'ébullition des alcanes et leurs densités augmentent avec l'augmentation du poids moléculaire. Tous les alcanes sont plus légers que l'eau, insolubles dans celle-ci, mais solubles dans les solvants non polaires (par exemple, dans le benzène) et sont eux-mêmes de bons solvants.
· Les points de fusion et d'ébullition diminuent du moins ramifié au plus ramifié.
Les alcanes gazeux brûlent avec une flamme incolore ou bleu pâle, dégageant de grandes quantités de chaleur.
La rotation des atomes autour de la liaison s ne la cassera pas. En raison de la rotation intramoléculaire le long des liaisons s C–C, les molécules d'alcane, à partir de l'éthane C 2 H 6 , peuvent prendre différentes formes géométriques.
Diverses formes spatiales d'une molécule, passant l'une dans l'autre par rotation autour de liaisons s C – C, sont appelées conformations ou isomères de rotation(conformes).
Les isomères de rotation d'une molécule sont ses états énergétiquement inégaux. Leur interconversion se produit rapidement et constamment à la suite d'un mouvement thermique. Par conséquent, les isomères de rotation ne peuvent pas être isolés individuellement, mais leur existence a été prouvée par des méthodes physiques.
alcanes
.
méthane, éthane, propane, butane
–fr
9. Hydrocarbures. Classification. Limiter les hydrocarbures de la série méthane. série homologue. Nomenclature. Isomérie. Radicaux. sources naturelles. Synthèse Fischer-Tropsch. Méthodes de préparation (à partir d'alcènes, d'acides carboxyliques, de dérivés halogénés, par la réaction de Wurtz)
Nom général (générique) des hydrocarbures saturés - alcanes
.
Les noms des quatre premiers membres de la série homologue du méthane sont triviaux : méthane, éthane, propane, butane
. A partir du cinquième nom, ils sont formés à partir de chiffres grecs avec l'ajout d'un suffixe –fr
Les radicaux (radicaux hydrocarbonés) ont également leur propre nomenclature. Les radicaux monovalents sont appelés alkyles
et sont désignés par la lettre R ou Alk.
Leur formule générale est C n H 2n+ 1 .
Les noms des radicaux sont formés à partir des noms des hydrocarbures correspondants en remplaçant le suffixe -une suffixer -limon(méthane - méthyle, éthane - éthyle, propane - propyle, etc.).
Les radicaux divalents sont nommés en changeant le suffixe -une sur le -ylidène(une exception est le radical méthylène == CH 2).
Les radicaux trivalents ont le suffixe -ylidine
Isomérie. Les alcanes sont caractérisés par une isomérie structurale. Si une molécule d'alcane contient plus de trois atomes de carbone, l'ordre de leur connexion peut être différent. Un des isomères du butane ( n-butane) contient une chaîne carbonée non ramifiée, et l'autre - isobutane - ramifiée (isostructure).
La source la plus importante d'alcanes dans la nature est le gaz naturel, les matières premières d'hydrocarbures minéraux - le pétrole et les gaz de pétrole associés.
La production d'alcanes peut être réalisée par la réaction de Wurtz, qui consiste en l'action du sodium métallique sur des dérivés monohalogénés d'hydrocarbures.
2CH 3 -CH 2 Br (bromure d'éthyle) + 2Na -> CH 3 -CH 2 -CH 2 -CH 3 (butane) + 2NaBr
des alcènes
C n H 2n + H 2 → C n H 2n+2
Synthèse Fischer-Tropsch
nCO + (2n+1)H 2 → C n H 2n+2 + nH 2 O
Le tableau montre que ces hydrocarbures diffèrent les uns des autres par le nombre de groupes -CH2- Une telle série de structures similaires, ayant des propriétés chimiques similaires et différant les unes des autres par le nombre de ces groupes est appelée série homologue. Et les substances qui le composent sont appelées homologues.
| Formule | Nom |
| CH 4 | méthane |
| C 2 H 6 | éthane |
| C 3 H 8 | propane |
| C 4 H 10 | butane |
| C 4 H 10 | isobutane |
| C 5 H 12 | pentane |
| C 5 H 12 | isopentane |
| C 5 H 12 | néopentane |
| C 6 H 14 | hexane |
| C 7 H 16 | heptane |
| C 10 H 22 | doyen |
10. Limiter les hydrocarbures (alcanes). Propriétés chimiques et physiques : réactions de substitution radicalaire. Halogénation, nitruration, sulfochloration, sulfoxydation. Le concept de réactions en chaîne.
Propriétés physiques
Dans des conditions normales, les quatre premiers membres de la série homologue des alcanes sont des gaz, C 5 -C 17 sont des liquides et à partir de C 18 sont des solides. Les points de fusion et d'ébullition des alcanes et leurs densités augmentent avec l'augmentation du poids moléculaire. Tous les alcanes sont plus légers que l'eau, insolubles dans celle-ci, mais solubles dans les solvants non polaires (par exemple, dans le benzène) et sont eux-mêmes de bons solvants.
Les acides et les bases ne présentent leurs propriétés qu'en présence les uns des autres.Pas une seule substance ne donnera un proton s'il n'y a pas d'accepteur de proton - une base dans le système, et vice versa. elles forment couple acide-base conjugué dans lequel plus l'acide est fort, plus sa base conjuguée est faible, et plus la base est forte, plus son acide conjugué est faible.
Un acide donne un proton pour devenir une base conjuguée, et une base accepte un proton pour devenir un acide conjugué. L'acide est généralement noté AN et la base B.
Par exemple : HC1- H++ + C1 -, HC1 est un acide fort ; C1 - ion - base faible conjuguée ;
CH 3 COOH - CH 3 COO - + H +, CH 3 COOH est un acide faible et CH 3 COO - est une base forte conjuguée à un ion.
La vue générale peut être représentée comme suit :
H+¦ : A + B H :B+ + A :-
les bases to-ta résistent. résister.
les bases de to-ta
Nous avons déjà dit que les propriétés acides des composés ne se trouvent qu'en présence d'une base, et les propriétés basiques - en présence d'un acide, c'est-à-dire dans les composés, il existe un certain équilibre acido-basique, pour l'étude duquel H 2 O est utilisé comme solvant. En ce qui concerne H 2 O en tant qu'acide ou en tant que base, les propriétés acido-basiques des composés sont déterminées.
Pour les électrolytes faibles, l'acidité est quantifiée À égal une réaction qui consiste en le transfert de H + d'un acide vers H 2 O en tant que base.
CH 3 COOH + H 2 O - CH 3 COO - + H 3 O +
à cet acide de base basique
CH 3 COO - - ion acétate, base conjuguée;
H 3 O + - ion hydronium, acide conjugué.
En utilisant la valeur de la constante d'équilibre de cette réaction et en tenant compte du fait que la concentration en H 2 O est pratiquement constante, il est possible de déterminer le produit K? appelée constante d'acidité À acidité (K une).

Plus il y a de K a, plus l'acide est fort. Pour CH 3 COOH K a \u003d 1,75 10 -5. ces petites valeurs ne sont pas pratiques dans les travaux pratiques, donc K a est exprimé par RK une (рК = -?g К une). Pour CH 3 COOH pKa = 4,75. Plus la valeur de pKa est petite, plus l'acide est fort.
La force des bases est déterminée par la valeur de pK ВН +.
Propriétés acides des composés organiques à groupements fonctionnels hydrogénés (alcools, phénols, thiols, acides carboxyliques, amines).
acides organiques
Dans les composés organiques, selon la nature de l'élément auquel H+ est associé, on distingue les acides suivants :
IL- acides (acides carboxyliques, phénols, alcools)
CH- acides (hydrocarbures et leurs dérivés)
NH- acides (amines, amides, imides)
SH- acides (thiols).
Un centre acide est un élément et son atome d'hydrogène associé.
La force de l'acide dépendra de stabilité des anions, celles. de la base conjuguée, qui se forme lorsque H + se détache de la molécule. Plus l'anion est stable, plus l'acidité du composé est élevée.
La stabilité de l'anion dépend d'un certain nombre de facteurs qui contribuent à la délocalisation de la charge. Plus la délocalisation de charge est élevée, plus l'anion est stable, plus les propriétés acides sont fortes.
Facteurs affectant le degré de délocalisation :
- 1. Nature de l'hétéroatome dans le centre acide
- 2. Effets électroniques des atomes de radicaux hydrocarbonés et de leurs substituants
- 3. La capacité des anions à se solvater.
- 1. Dépendance de l'acidité vis-à-vis de l'hétéroatome.
La nature d'un hétéroatome est comprise comme son électronégativité (E.O.) et sa polarisabilité. Plus il y a de (E.O.), plus l'écart hétérolytique dans la molécule est facile à réaliser. Dans les périodes de gauche à droite, avec une augmentation de la charge du noyau, (E.O) augmente, c'est-à-dire la capacité des éléments à retenir une charge négative. En raison du déplacement de la densité électronique, la liaison entre les atomes est polarisée. Plus il y a d'électrons et plus le rayon de l'atome est grand, plus les électrons du niveau d'énergie externe sont éloignés du noyau, plus la polarisabilité est élevée et plus l'acidité est élevée.
Exemple : CH- NH- OH- SH-
augmentation de l'E.O. et acidité
C, N, O - éléments de la même période. E.O. augmente avec le temps, l'acidité augmente. Dans ce cas, la polarisabilité n'affectera pas l'acidité.
La polarisabilité des atomes dans la période varie légèrement, par conséquent, le principal facteur déterminant l'acidité est l'E.O.
Considérons maintenant OH-SH-
augmentation de l'acidité
O, S - sont dans le même groupe, le rayon dans le groupe augmente de haut en bas, par conséquent, la polarisabilité de l'atome augmente également, ce qui entraîne une augmentation de l'acidité. S a un rayon atomique plus grand que O, de sorte que les thiols présentent des propriétés acides plus fortes que les alcools.
Comparez trois composés : éthanol, éthanethiol et aminoéthanol :
H3C-CH2- IL, H3C-CH2- SH et H3C-CH2- NH 2
- 1. Comparez par radical - ils sont identiques;
- 2. Par la nature de l'hétéroatome dans le groupe fonctionnel : S et O sont dans le même groupe, mais S a un rayon atomique plus grand, une polarisabilité plus élevée, donc l'éthanethiol a des propriétés acides plus fortes
- 3. Comparons maintenant O et N. O a un EO plus élevé, donc l'acidité des alcools sera plus élevée.
- 2. Influence du radical hydrocarboné et de ses substituants
Il faut attirer l'attention des élèves sur le fait que les composés comparés doivent avoir le même centre acide et le même solvant.
Substituants électroattracteurs (EA) contribuent à la délocalisation de la densité électronique, ce qui conduit à la stabilité de l'anion et, par conséquent, à une augmentation de l'acidité.
Substituants donneurs d'électrons (ED) au contraire, ils contribuent à la concentration de la densité électronique dans le centre acide, ce qui entraîne une diminution de l'acidité et une augmentation de la basicité.
Par exemple : les alcools monohydriques présentent des propriétés acides plus faibles que les phénols.
Exemple : H 3 C > CH 2 > OH
- 1. Le centre acide est le même
- 2. Le solvant est le même
Dans les alcools monovalents, la densité électronique passe du radical hydrocarbure au groupe OH, c'est-à-dire le radical présente un effet + I, puis une grande quantité de densité électronique est concentrée sur le groupe OH, à la suite de quoi H + est plus fermement lié à O et il est difficile de rompre la liaison OH, par conséquent, les alcools monohydriques présentent une faible propriétés acides.
Dans le phénol, au contraire, le cycle benzénique est E.A. et le groupe OH est E.D.
En raison du fait que le groupe hydroxyle est inclus dans la conjugaison p-p commune avec le cycle benzénique, une délocalisation de la densité électronique se produit dans la molécule de phénol et l'acidité augmente, tk. la conjugaison s'accompagne toujours d'une augmentation des propriétés acides.
Une augmentation du radical hydrocarbure dans les acides monocarboxyliques affecte également le changement des propriétés acides, et lorsque des substituants sont introduits dans l'hydrocarbure, les propriétés acides changent.
Exemple: dans les acides carboxyliques, lors de la dissociation, des ions carboxylate se forment - les anions organiques les plus stables.

Dans l'ion carboxylate, la charge négative due à la conjugaison p, p est répartie de manière égale entre deux atomes d'oxygène, c'est-à-dire il est délocalisé et, par conséquent, moins concentré; par conséquent, le centre acide des acides carboxyliques est plus fort que celui des alcools et des phénols.



Avec une augmentation du radical hydrocarbure, qui joue le rôle de E.D. l'acidité des acides monocarboxyliques diminue en raison d'une diminution de q + sur l'atome de carbone du groupe carboxyle. Par conséquent, dans la série homologue des acides, l'acide formique est le plus fort.



Avec l'introduction d'E.A. substituant dans un radical hydrocarboné, tel que le chlore - l'acidité du composé augmente, car en raison de l'effet -I, la densité électronique est délocalisée et q + sur l'atome C du groupe carboxyle augmente, donc, dans cet exemple, l'acide trichloroacétique sera le plus fort.
3. Influence du solvant.
L'interaction des molécules ou des ions d'un soluté avec un solvant est appelée un processus solvatation. La stabilité d'un anion dépend essentiellement de sa solvatation en solution : plus l'ion est solvaté, plus il est stable, et plus la solvatation est importante, plus la taille de l'ion est petite et moins la charge négative y est délocalisée.
Conférence #4
Conférence #4
ACIDES ET BASES ORGANIQUES
- Théorie des protons des acides et des bases de Bronsted.
- Classification des acides et des bases selon Bronsted.
- Influence des facteurs structuraux sur l'acidité et la basicité.
- Acides et bases de Lewis. La théorie des acides et bases durs et mous.
Il existe actuellement deux principaux
théories des acides et des bases : la théorie de Bronsted et la théorie de Lewis.
Théorie des protons des acides et
terrain de Bronsted
Acides bronzés - euh puis les connexions
capable de donner un proton (donneurs de protons).
Fondation de Bronsted - sont des composés qui peuvent accepter un proton
(accepteurs de protons). Pour interagir avec un proton, la base doit avoir
une paire libre d'électrons ou d'électrons de liaison p.
Les acides et les bases forment des conjugués
des paires acide-base, par exemple :
En général :
La force de l'acide HA dépendra de la force de la base
:V. Par conséquent, pour créer une échelle unifiée, la force des acides et des bases de Bronsted
déterminé par rapport à l'eau, qui est un composé amphotère et peut
présentent à la fois des propriétés acides et basiques.
La force des acides est déterminée par la constante d'équilibre
leurs interactions avec l'eau comme base, par exemple :
CH 3 COOH + H 2 O CH 3 COO - + H 3 O +
.gif)
Puisque dans des solutions diluées
=const, alors il peut être ajouté à
constante d'équilibre, appelée constante d'acidité :
.gif)
En pratique, les valeurs sont souvent utilisées
paquet une = - lg K une . Comment
moins la valeur pK une, le plus fort
acide.
La force des bases est déterminée par la constante
équilibre de leur interaction avec l'eau en tant qu'acide :
RNH 2 + H 2 O RNH 3 + + OH -
.gif) —
—
constante de basicité.
Pour acides et bases conjugués
K a K b =K W . Ainsi, dans
couple acide-base conjugué, plus l'acide est fort, plus la base est faible et
vice versa. La force de la base est souvent exprimée non pas par la constante de basicité, mais par la constante
acidité de l'acide conjugué.
Par exemple, pour la base RNH 2 l'ampleur est
acide conjugué constante d'acidité:
RNH 3 + + H 2 O RNH 2 + H 3 O +
En pratique, la valeur est souvent utilisée ![]() . Plus la valeur est grande, plus
. Plus la valeur est grande, plus
base plus solide.
Classement biologique
acides et bases
Les acides et les bases de Bronsted sont classés selon
la nature de l'atome à un centre acide ou basique.
Selon la nature de l'élément avec lequel
lié au proton, il existe quatre principaux types d'acides organiques
Bronsted :
- O-H - acides- acides carboxyliques,
alcools, phénols; - S-H - acides- les thiols ;
- N-H - acides- amines, amides,
imides; - C-H - acides— les hydrocarbures et leurs
dérivés.
En fonction de la
la nature de l'atome, à la seule paire d'électrons dont un proton est attaché,
Les fondations Bronsted sont divisées en trois types principaux:
- bases ammoniacales- les amines,
les nitriles, les composés hétérocycliques azotés ; - bases d'oxonium- les alcools,
éthers, aldéhydes, cétones, acides carboxyliques et leurs fonctions
dérivés; - bases de sulfonium- les thiols,
sulfures.
type spécial
Les terrains de Bronsted représentent p - bases dont le centre de basicité est
électrons p - communication
(alcènes, arènes).
Influence des facteurs structurels sur
force relative des acides et des bases
La force d'un acide ou d'une base est déterminée
la position d'équilibre de l'interaction acide-base et dépend de la différence
énergies libres des composés initiaux et finaux. Par conséquent, les facteurs qui
stabiliser davantage la base conjuguée que l'acide, augmenter
l'acidité et réduire la basicité. Facteurs stabilisant principalement
un acide par rapport à une base agit dans le sens opposé.
Puisque les bases conjuguées portent généralement une charge négative, alors
les facteurs de stabilisation des anions contribuent à une augmentation de l'acidité.
L'effet de la structure sur la force des acides et
terrains.
Acides bronzés.
La force d'un acide dépend de la nature de l'atome à
centre acide et son environnement structural.
Pour évaluer la force relative des acides, les éléments suivants sont importants :
caractéristiques d'un atome à un centre acide comme son électronégativité et
polarisabilité.
Toutes choses égales par ailleurs, pour des éléments de même
période avec une augmentation de l'électronégativité de l'atome, l'acidité des composés
augmente, car la forte électronégativité de l'atome au centre acide
stabilise l'anion formé lors de l'élimination d'un proton. Oui, l'acidité.
diminue dans la série :
Acides OH> Acides NH>
CH-acides
| CH 3 O-H |
CH 3 NH-H |
CH3CH2-H |
|
| pKa |
16 |
30 |
40 |
L'électronégativité d'un atome ne dépend pas seulement de
de sa nature, mais aussi du type d'hybridation et augmente avec l'augmentation
caractère s des orbitales hybrides. Dans le même temps, l'acidité augmente
Connexions:
L'augmentation de l'acidité des composés, malgré
une diminution de l'électronégativité des atomes d'un sous-groupe est associée à une augmentation de leur
polarisabilité à mesure que le rayon de l'atome augmente. Grande polarisabilité de l'atome
contribue à une meilleure délocalisation de la charge négative et à une stabilité accrue
base conjuguée.
Avec la même nature de l'atome avec l'acide
au centre, la force d'un acide est déterminée par son environnement structurel. Augmentation de la force
l'acide favorise la délocalisation de la charge négative dans la base conjuguée
(anion) et sa dispersion sur plusieurs atomes.
Ainsi, les acides carboxyliques sont l'un des plus forts
acides organiques. Leur force est due à la stabilisation de l'anion carboxylate pour
délocalisation de la charge négative dans le système conjugué. Par conséquent
la charge négative dans l'anion carboxylate est dispersée entre deux atomes
l'oxygène, et les deux liaisons C-O sont absolument équivalentes :
.gif)
Les phénols sont des acides plus forts que
alcools, en raison de la stabilisation par résonance de l'anion phénolate, charge négative
qui est délocalisé le long du cycle aromatique :
.gif)
En conséquence, la force des acides OH organiques
peuvent être placés dans l'ordre suivant :
| ROH |
| H2O |
| ArOH |
| RCOOH |
|
| pKa |
16-17 |
15,7 |
8-11 |
4-5 |
L'introduction d'un substituant dans la liaison acide
le centre du radical hydrocarbure affecte la force de l'acide. attracteur d'électrons
les substituants augmentent et les donneurs d'électrons - réduisent l'acidité. Influence
substituants attracteurs d'électrons est lié à leur capacité à délocaliser
charges négatives et plus
stabiliser la base conjuguée (anion). Effet du donneur d'électrons
substituants, au contraire, conduit à la déstabilisation de l'anion.
Augmentation des substituants attracteurs d'électrons
force des acides carboxyliques aliphatiques et aromatiques, donneur d'électrons
les substituants agissent en sens inverse :
| Cl-CH 2 -COOH |
H-COOH |
CH 3 -COOH |
|
| pKa |
2,8 |
3,7 |
4,7 |
.gif) |
.gif) |
.gif) |
|
| +M > -I |
-M et -I |
||
| pKa | 4,47 |
4,20 |
3,43 |
Les substituants ont un effet similaire sur
acidité des alcools et des phénols.
Fondation de Bronsted.
Avec le même environnement structurel pour
éléments de la même période avec une augmentation de l'électronégativité de l'atome au niveau principal
le centre de la basicité des composés diminue :
bases ammonium > bases oxonium Je suis
| ROH |
RNH 2 |
|
| |
~2 |
~10 |
La diminution de la basicité est due au fait que plus
un atome électronégatif maintient plus fermement la seule paire d'électrons,
qu'il doit donner pour former une liaison avec le proton.
Augmentation du caractère s des orbitales hybrides
conduit à une diminution de la basicité :
.gif)
Pour les éléments d'un sous-groupe avec des
la basicité de la charge de base diminue :
bases oxonium > sulfonium
terrains
Introduction de substituants donneurs d'électrons
augmente, et l'introduction de l'électroaccepteur - abaisse la basicité. Alors,
les substituants donneurs d'électrons augmentent la basicité des composés aliphatiques et
amines aromatiques, augmentant la propension de la paire d'électrons d'azote à attaquer
proton. Les substituants attracteurs d'électrons, au contraire, réduisent la densité électronique
seule paire d'électrons d'azote et la rend moins susceptible d'être attaquée
proton:
| |
9,2 |
10,6 |
10,7 |
Si une paire libre d'électrons d'azote est dans
conjugaison avec une double liaison ou un cycle aromatique, la basicité est réduite.
Ainsi, dans l'aniline, une paire libre d'électrons d'azote est conjuguée à un aromatique
bague.
La protonation de l'aniline conduit à une violation
conjugaison et est énergétiquement moins favorable que la protonation des aliphatiques
amines.
.gif) |
|||
| |
10,6 |
4,6 |
0,9 |
Les amides d'acides carboxyliques sont très faibles
bases dues à la conjugaison d'une paire d'électrons d'azote avec un groupe carbonyle. V
En conséquence, l'atome d'azote acquiert un positif partiel et l'atome d'oxygène -
charge négative partielle, et la protonation des amides se produit, en règle générale,
par atome d'oxygène.
.gif)
Basicité des hétérocycles azotés
composés est également déterminée par la disponibilité d'une paire d'électrons d'azote pour attaquer
proton. Les hétérocycles contenant de l'azote saturé ont une basicité élevée, en
dans lequel l'atome d'azote est à l'état sp 3 -hybridation. Basicité de l'atome d'azote du pyridinium
(sp 2 hybridation) ci-dessous. Enfin,
l'atome d'azote du pyrrole est pratiquement dépourvu de propriétés basiques, puisque son
protonation désigne la destruction d'un hétérocycle aromatique
systèmes :
| |
|
|
|
| pKa |
11,27 |
5,2 |
— 0.3 |
Acides et bases
Lewis
J. Lewis a proposé une théorie plus générale
acides et bases.
Fondations Lewis ce sont les donateurs du couple
électrons (alcools, anions alcoolates, éthers, amines, etc.)
Acides de Lewis - ce sont les accepteurs de la paire
électrons,
celles. composés qui ont
orbitale vacante (ion hydrogène et cations métalliques : H + ,
Ag + , Na + , Fe 2+ ;
halogénures d'éléments des deuxième et troisième périodes BF 3 ,
AlCl 3 , FeCl 3 , ZnCl 2 ; halogènes; composés d'étain et de soufre :
SnCl 4, SO3).
Ainsi, les fondements de Bronsted et Lewis sont -
ce sont les mêmes particules. Cependant, selon Bronsted, la basicité est la capacité
n'attachent qu'un proton, tandis que la basicité de Lewis est plus
large et signifie la capacité d'interagir avec toute particule ayant
orbitale libre basse.
L'interaction acide-base de Lewis est
l'interaction donneur-accepteur et toute réaction hétérolytique peuvent être
représentent comme l'interaction d'un acide de Lewis et d'une base de Lewis :
.gif)
Une seule échelle pour comparer la force des acides et
Les bases de Lewis n'existent pas, puisque leur force relative dépendra de
quelle substance est prise comme norme (pour les acides et les bases de Bronsted tels
l'eau est la norme). Pour évaluer la facilité d'écoulement de l'acide-base
interaction selon Lewis R. Pearson a proposé une théorie qualitative
acides et bases « durs » et « mous ».
Bases rigides avoir un haut
électronégativité et faible polarisabilité. Ils sont difficiles à oxyder. Leur
les orbitales moléculaires occupées les plus élevées (HOMO) ont une faible énergie.
Sols mous avoir peu
électronégativité et haute polarisabilité. Ils s'oxydent facilement. Leur plus haut
les orbitales moléculaires occupées (HOMO) ont une énergie élevée.
Acides durs avoir un haut
électronégativité et faible polarisabilité. Ils sont difficiles à récupérer. Leur
les orbitales moléculaires libres les plus basses (LUMO) ont une faible énergie.
Acides doux avoir un faible
électronégativité et haute polarisabilité. Ils sont faciles à récupérer.
Leurs orbitales moléculaires libres les plus basses (LUMO) sont à haute énergie.
L'acide le plus dur
H + , le plus doux
CH 3Hg + . Plus
bases rigides - F- et
Oh- , le plus doux
je- et n - .
Tableau 5. Acides durs et mous
et fondations.
| Rigide |
Intermédiaire |
Mou, tendre |
| acides |
||
| H+, Na+, K+, Mg 2+ , Ca 2+ , Al 3+ , Fe 3+ , BF 3 , AlCl 3 , RC + =O |
Cu 2+, Fe 2+, Zn 2+ , R 3 C + |
Ag + , Hg 2+ , je 2 |
| Fondations |
||
| H 2 O, OH - , F - , ROH, RO-, R2O, NH3, RNH2 |
ArNH 2 , Br -, C5H5N |
R2S, RSH, RS-, I-, H-, C 2 H 4 , C 6 H 6 |
Principe des acides et bases durs et mous
Pearson (principe GIC):
Acides durs principalement
réagir avec des bases dures et des acides mous avec des
terrains.
Cela se traduit par des vitesses de réaction élevées et par
la formation de composés plus stables, puisque l'interaction entre des
les orbitales sont plus économes en énergie que l'interaction entre les orbitales,
sensiblement différente en énergie.
Le principe GMLC est utilisé pour déterminer
sens prédominant des processus concurrents (réactions d'élimination et
substitution nucléophile, réactions impliquant des nucléophiles ambiants) ; pour
création ciblée de détoxifiants et de médicaments.
Selon la théorie de Lowry-Bronsted, les acides sont des substances capables de donner un proton, les bases sont des substances qui acceptent un proton :
Si B est une base forte, alors c'est un acide faible. Avec de l'aide, vous pouvez caractériser le degré de dissociation d'un acide ou d'un acide conjugué. Outre la constante d'acidité, il existe également le concept de constante de basicité et la constante correspondante.
![]()
Selon la théorie de Lewis, les acides sont des composés qui peuvent accepter, les bases peuvent donner une paire d'électrons.
Au sens large, les acides sont des composés qui fournissent un cation, dans un cas particulier, un proton, ou acceptent une paire d'électrons avec un atome ou un groupe d'atomes, etc.).
Les bases acceptent un cation, dans un cas particulier, un proton, ou fournissent une paire d'électrons avec un atome ou un groupe d'atomes
L'acidité ou la basicité d'une substance se manifeste dans le processus d'interaction avec une autre substance, en particulier avec un solvant, et est donc relative.
De nombreuses substances ont des propriétés amphotères. Par exemple, l'eau, les alcools et les acides sont capables de donner un proton lorsqu'ils interagissent avec des bases et de l'accepter avec des acides. En l'absence d'acides et de bases, la double nature de tels composés se manifeste par l'autoprotolyse :
La dissociation d'un acide dans un solvant signifie le transfert d'un proton vers le solvant :
![]()
A cet égard, la force de l'acide est exprimée par la constante de dissociation, qui n'est caractéristique que pour un solvant donné. Le transfert de protons ne se produit que dans des solvants hautement ionisants et solvatants, tels que l'eau.
Le degré de dissociation acide lors de la transition d'un milieu aqueux à un milieu organique diminue de 4 à 6 ordres de grandeur.
Les solvants fortement solvatants et ionisants neutralisent la force des acides, tandis que les solvants non polaires et faiblement iolaires, interagissant avec eux au niveau des liaisons hydrogène, ont un effet différenciateur. Dans ce dernier cas, les différences de force des acides deviennent plus importantes.
Dans les solvants inertes et non polaires, la probabilité de détachement de protons est très faible, bien qu'en raison d'effets électroniques internes, la liaison puisse être fortement polarisée. Dans de telles conditions, les propriétés acides se manifestent par l'auto-association de molécules HA ou en association avec des accepteurs de protons, des bases. Dans ce dernier cas, la mesure de l'acidité est la constante d'association avec une base choisie comme étalon. Par exemple, la constante d'association de l'acide benzoïque et de la diphénylguanidine dans le benzène est
Le pouvoir protonisant d'un acide s'exprime également en fonction de la fonction acidité, qui caractérise l'état d'équilibre lors de la complexation des acides et des bases dans les solvants organiques. Les bases les plus couramment utilisées sont des indicateurs qui changent de couleur en fonction de la force de l'acide, ce qui permet d'étudier le système par des méthodes spectroscopiques. Dans ce cas, il est important que les bandes de la base libre associée soient identifiées dans le spectre.
Ainsi, en milieu d'introduction, les acides et les bases forment des ions solvatés, en milieu organique, des paires d'ions et leurs associés.
Le concept de formation de complexes est proche du concept d'association : en raison des interactions donneur-accepteur et datif, des complexes électron-donneur-accepteur, également appelés complexes de transfert de charge, peuvent se former à partir d'ions et de molécules.
Types de donneurs d'électrons : I) composés à hétéroatomes. contenant des paires isolées d'électrons, des éthers, des amines, des sulfures, des iodures, etc. Par exemple : éther diéthylique otlampn. ldméthylsulfpd iodure de triphényl-phosphine-propyle
2) les composés contenant des liaisons -éthylène, acétylènes, benzène et ses dérivés, autres systèmes aromatiques ;
3) composés capables de transférer des électrons - liaisons alcanes, cycloalcanes :

Types d'accepteurs d'électrons : 1) composés métalliques contenant une orbitale vacante (orbitale K) : halogénures, etc., ions métalliques
2) composés capables d'accepter une paire d'électrons par halogènes anti-liants vacants, halogènes mixtes
3) composés avec des liaisons - avec des substituants fortement électronégatifs participant à la formation de complexes en raison du relâchement du tétracyanoéthylène trinitrobenzène
Ainsi, soit le donneur peut interagir avec l'accepteur vacant, formant un nouveau MO avec une diminution de l'énergie du système :

En chimie organique, les -complexes sont de la plus haute importance, et les -complexes sont caractérisés par des constantes d'instabilité, qui sont, en fait, leurs constantes de dissociation.
Les constantes de dissociation et d'association des acides et des bases ne décrivent pas encore complètement leurs propriétés. Un rôle important dans la compréhension de nombreux processus chimiques, et en particulier du phénomène de catalyse, a été joué par le concept d'acides et de bases durs et mous (le principe
GMCCO). Conformément à ce concept, les acides et les bases apparentés interagissent le plus efficacement : un acide doux avec une base molle, un acide dur avec une base dure.
Signes d'acides et de bases durs (tableau 8) : 1) petite taille d'un ion ou d'une molécule ; 2) haute électronégativité ; 3) charge localisée ; 4) faible polarisabilité ; 5) les orbitales vacantes les plus basses (HVO) des acides ont une énergie élevée ; 6) les orbitales les plus remplies (HOO) des bases ont une faible énergie.
Selon Lewis, les propriétés acides et basiques des composés organiques sont mesurées par la capacité d'accepter ou de donner une paire d'électrons, suivie de la formation d'une liaison. Un atome qui accepte une paire d'électrons est un accepteur d'électrons, et un composé contenant un tel atome doit être classé comme un acide. Un atome qui fournit une paire d'électrons est un donneur d'électrons, et un composé contenant un tel atome est une base.
Plus précisément, les acides de Lewis peuvent être un atome, une molécule ou un cation : proton, halogénures d'éléments des deuxième et troisième groupes du système périodique, halogénures de métaux de transition - BF3, ZnCl2, AlCl3, FeCl3, FeBr3, TiCl4, SnCl4, SbCl5, métal cations, anhydride sulfurique - SO3, carbocation. Les bases de Lewis comprennent les amines (RNH2, R2NH, R3N), les alcools ROH, les éthers ROR
Selon Bronsted-Lowry, les acides sont des substances capables de donner un proton et les bases sont des substances qui acceptent un proton.
Acide et base conjugués :
HCN (acide) et CN- (base)
NH3 (base) et NH4+ (acide)
L'équilibre acido-basique (ou protolytique) est un équilibre auquel participe un proton (H+).
HCOOH + H 2 O D H 3 O + + HCOO -
acide 2 base 1
H 2 O + NH 3 D NH 4 + + OH -.
acide 1 base 2 conjugué conjugué
acide 2 base 1
7. Types d'isomérie en chimie organique. Isomérie structurale, spatiale et optique. Chiralité. Compatibilité et configuration. R, S, Z, E - nomenclature.
Il existe deux types d'isomérie : structurelle et spatiale (stéréoisomérie). Les isomères structuraux diffèrent les uns des autres dans l'ordre des liaisons des atomes dans une molécule, les stéréo-isomères - dans la disposition des atomes dans l'espace avec le même ordre de liaisons entre eux.
Isomérie structurale : isomérie du squelette carboné, isomérie de position, isomérie de diverses classes de composés organiques (isomérie interclasse).
Isomérie structurale
Isomérie du squelette carboné
 L'isomérie de position est due à la position différente de la liaison multiple, du substituant, du groupe fonctionnel avec le même squelette carboné de la molécule :
L'isomérie de position est due à la position différente de la liaison multiple, du substituant, du groupe fonctionnel avec le même squelette carboné de la molécule :
Isomérie spatiale
L'isomérie spatiale est divisée en deux types : géométrique et optique.
 L'isomérie géométrique est caractéristique des composés contenant des doubles liaisons et des composés cycliques. La libre rotation des atomes autour d'une double liaison ou dans un cycle étant impossible, les substituants peuvent être situés soit d'un côté du plan de la double liaison ou du cycle (position cis), soit sur des côtés opposés (position trans).
L'isomérie géométrique est caractéristique des composés contenant des doubles liaisons et des composés cycliques. La libre rotation des atomes autour d'une double liaison ou dans un cycle étant impossible, les substituants peuvent être situés soit d'un côté du plan de la double liaison ou du cycle (position cis), soit sur des côtés opposés (position trans).

 L'isomérie optique se produit lorsqu'une molécule est incompatible avec son image dans un miroir. Ceci est possible lorsque l'atome de carbone de la molécule a quatre substituants différents. Cet atome est dit asymétrique.
L'isomérie optique se produit lorsqu'une molécule est incompatible avec son image dans un miroir. Ceci est possible lorsque l'atome de carbone de la molécule a quatre substituants différents. Cet atome est dit asymétrique.
CHIRALITÉ, propriété d'un objet d'être incompatible avec sa réflexion dans un miroir plan idéal.
Diverses structures spatiales qui surviennent en raison de la rotation autour de liaisons simples sans violer l'intégrité de la molécule (sans rompre les liaisons chimiques) sont appelées CONFORMATIONS.
La structure des alcanes. Sp3 est l'état du carbone. Caractérisation des liaisons C-C et C-H. Le principe de rotation libre. conformation. Modes de représentation et nomenclature. Propriétés physiques des alcanes.